- Research Article
- Open access
- Published:
A Bayesian Analysis for Identifying DNA Copy Number Variations Using a Compound Poisson Process
EURASIP Journal on Bioinformatics and Systems Biology volume 2010, Article number: 268513 (2010)
Abstract
To study chromosomal aberrations that may lead to cancer formation or genetic diseases, the array-based Comparative Genomic Hybridization (aCGH) technique is often used for detecting DNA copy number variants (CNVs). Various methods have been developed for gaining CNVs information based on aCGH data. However, most of these methods make use of the log-intensity ratios in aCGH data without taking advantage of other information such as the DNA probe (e.g., biomarker) positions/distances contained in the data. Motivated by the specific features of aCGH data, we developed a novel method that takes into account the estimation of a change point or locus of the CNV in aCGH data with its associated biomarker position on the chromosome using a compound Poisson process. We used a Bayesian approach to derive the posterior probability for the estimation of the CNV locus. To detect loci of multiple CNVs in the data, a sliding window process combined with our derived Bayesian posterior probability was proposed. To evaluate the performance of the method in the estimation of the CNV locus, we first performed simulation studies. Finally, we applied our approach to real data from aCGH experiments, demonstrating its applicability.
1. Introduction
Cancer progression, tumor formations, and many genetic diseases are related to aberrations in some chromosomal regions. Chromosomal aberrations are often reflected in DNA copy number changes, also known as copy number variations (CNVs) [1]. To study such chromosomal aberrations, experiments are often conducted based on tumor samples from a cell-line-using technologies such as aCGH or SNP arrays. For instance, in aCGH experiments, a DNA test sample and a diploid reference sample are first fluorescently labeled by Cy3 and Cy5. Then, the samples are mixed and hybridized to the microarray. Finally, the image intensities from the test and reference samples can be obtained for all DNA probes (bio-markers) along the chromosome [2, 3]. The log-base-2 ratios of the test and reference intensities, usually denoted as  , are used to generate an aCGH profile [4]. To reduce noise, the Gaussian-smoothed profile is often used. With an appropriate normalization process,
, are used to generate an aCGH profile [4]. To reduce noise, the Gaussian-smoothed profile is often used. With an appropriate normalization process,  is viewed as a Gaussian distribution of mean 0 and variance
is viewed as a Gaussian distribution of mean 0 and variance  [4, 5]. The deviation from mean 0 and variance
[4, 5]. The deviation from mean 0 and variance  in
in  data may indicate a copy number change. Therefore, detecting DNA copy number changes becomes the problem of how to identify significant parameter changes occurred in the sequence of
data may indicate a copy number change. Therefore, detecting DNA copy number changes becomes the problem of how to identify significant parameter changes occurred in the sequence of  observations.
observations.
There are a number of computational and statistical methods developed for the detection of CNVs based on aCGH data and SNP data. Examples include a finite Gaussian mixture model [6], pair wise t-tests [7], adaptive weights smoothing [8], circular binary segmentation (CBS) [4], hidden Markov modeling (HMM) [9], maximum likelihood estimation [10], and many others. A comparison between several of these methods for the analysis of aCGH data was given by Lai et al. [11]. There are continued efforts on developing methods for accurate detection of CNVs. Nannya et al. [12] developed a robust algorithm for copy number analysis of the human genome using high-density oligonucleotide microarrays. Price et al. [13] adapted the Smith-Waterman dynamic programming algorithm to provide a sensitive and robust approach (SW-ARRAY). More recently, Shah et al. [14] proposed a simple modification to the hidden Markov model (HMM) to make it be robust to outliers in aCGH data. Yu et al. [15] developed an edge detection algorithm for copy number analysis in SNP data. An algorithm called reversible jump aCGH (RJaCGH) for identifying copy number alterations was introduced in Rueda and Díaz-Uriarte [16]. This RJaCGH algorithm is based on a nonhomogeneous HMM fitted by reversible jump MCMC using Bayesian approach. Pique-Regi et al. [17] proposed to use piecewise constant (PWC) vectors to represent genome copy number and used sparse Bayesian learning (SBL) to detect copy number alterations breakpoints. Rancoita et al. [18] provided an improved Bayesian regression method for data that are noisy observations of a piecewise constant function and used this method for CNV analysis. We have formulated the problem as a statistical change-point detection [19] and proposed a mean and variance change-point model (MVCM), which brought significant improvement over many existing methods such as the CBS proposed by Olshen et al. [4].
The above-mentioned algorithms, however, have not taken advantage of other information such as the positions of the DNA probes or biomarkers along the chromosome. Recently, many researchers have begun to consider variations in the distance between biomarkers, gene density, and genomic features in the process of identifying increased or decreased chromosomal region of gene expression [5]. Several notable methods emerged along this line and we list a few of them here. Levin et al. [5] developed a scan statistics for detecting spatial clusters of genes on a chromosome based on gene positions and gene expression data modeled by a compound Poisson process on the basis of two independent simple Poisson processes. Daruwala et al. [20] developed a statistical algorithm for the detection of genomic aberrations in human cancer cell lines, where the location of aberrations in the copy numbers was modeled by a Poisson process. They distinguished genes as "regular" and "deviated", where the regular genes refer to those that have not been affected by chromosomal aberrations while the deviated genes are those whose log-transformed expression follows a Gaussian distribution with unknown mean and variance [20]. Sun et al. [21] developed a SNP association scan statistic similar to that of Levin et al. [5] using a compound Poisson process, which considers the complex distribution of genome variations in chromosomal regions with significant clusters of SNP associations.
Improvements have been made with the above more sophisticated modeling of the aCGH using both the log-intensity ratios and biomarker positions. The computation involved in this type of modeling is usually demanding and further improvement is needed. Motivated by these existing works, we propose to use a compound Poisson process approach to model the genomic features in identifying chromosomal aberrations. We use a Bayesian approach to determine an aberration (or a change-point) in the aCGH profile modeled by a compound Poisson process. In our model, the occurrences of the biomarkers are modeled by a homogeneous Poisson process and the aCGH is modeled by a Gaussian distribution. This novel method is able to identify the aberration corresponding to the CNVs with associated distance between biomarkers on the chromosome. The proposed method is inspired by the scan statistic [5, 21], which is widely used for identifying chromosomal aberrations. However, our method differs from the work of Levin et al. [5] in that our method uses a statistical change-point model with a compound Poisson process for the identification of CNVs.
2. Methods
2.1. Modeling aCGH Data Using a Compound Poisson Change Point Model
To describe our approach, we first describe a change-point model for a compound Poisson process in terms of the normalized log ratio  and the biomarker distances along a chromosome, where
and the biomarker distances along a chromosome, where  and
and  and
and  are the intensities of the test and reference samples at locus
are the intensities of the test and reference samples at locus  on the chromosome (or genome). Based on probability distribution theories and characteristics of the hybridization process of aCGH technique, the occurrence of the biomarkers on the chromosome can be modeled by a homogeneous Poisson process. Similarly to the notations adopted in Levin et al. [5] and Sun et al. [21], we denote
on the chromosome (or genome). Based on probability distribution theories and characteristics of the hybridization process of aCGH technique, the occurrence of the biomarkers on the chromosome can be modeled by a homogeneous Poisson process. Similarly to the notations adopted in Levin et al. [5] and Sun et al. [21], we denote  as a simple (homogeneous) Poisson process with the rate parameter
as a simple (homogeneous) Poisson process with the rate parameter  , where
, where  is the number of biomarkers occurring over a given base pair length
is the number of biomarkers occurring over a given base pair length  and
and  is the occurrence rate of biomarkers over a distance of t base pairs along the chromosome. Let
is the occurrence rate of biomarkers over a distance of t base pairs along the chromosome. Let  represent the positions of the biomarkers on a chromosome and
represent the positions of the biomarkers on a chromosome and

represent the distance between the  th biomarker and the
th biomarker and the  th biomarker. Since
th biomarker. Since  is a homogeneous Poisson process, according to probability distribution theories,
is a homogeneous Poisson process, according to probability distribution theories,  s are independent and identically distributed (iid) with exponential variables with parameter
s are independent and identically distributed (iid) with exponential variables with parameter  ; furthermore,
; furthermore,  s are gamma distributed with rate parameter
s are gamma distributed with rate parameter  and scale parameter i, and the probability density function as follows:
and scale parameter i, and the probability density function as follows:

where  is the gamma function, and
is the gamma function, and  for a positive integer
for a positive integer  .
.
Note the fact that the distances  s are iid exponential random variables can be used to verify the assumption on the occurrence of
s are iid exponential random variables can be used to verify the assumption on the occurrence of  being a simple (homogeneous) Poisson process.
being a simple (homogeneous) Poisson process.
Assume that the given interval with base pair length  is divided by the nonoverlapping subintervals with lengths
is divided by the nonoverlapping subintervals with lengths  . Then, the sequence of the log intensity ratio,
. Then, the sequence of the log intensity ratio,  , corresponding to each subinterval can be denoted as
, corresponding to each subinterval can be denoted as  and clearly
and clearly

Given that  is a homogeneous Poisson process and
is a homogeneous Poisson process and  follow independent Gaussian (normal) distributions [5] with mean
follow independent Gaussian (normal) distributions [5] with mean  and variance
and variance  ,
,  ,
,  is then defined by a compound Poisson process, where the
is then defined by a compound Poisson process, where the  ,
,  are independently and normally distributed with mean
are independently and normally distributed with mean  and variance
and variance  , respectively. The number,
, respectively. The number,  , of biomarkers in each subinterval of length
, of biomarkers in each subinterval of length  is distributed as a Poisson distribution with parameter
is distributed as a Poisson distribution with parameter  (where
(where  represents the occurrence rate of biomarkers or SNPs corresponding to subinterval
represents the occurrence rate of biomarkers or SNPs corresponding to subinterval  ) for
) for  .
.
The problem is if there is an aberration (increase or decrease) in the sequence  at an unknown locus
at an unknown locus  with base pair length
with base pair length  . In statistical change-point modeling theory, this is to know if there is a change in the parameters of the distribution of the independent sequence of
. In statistical change-point modeling theory, this is to know if there is a change in the parameters of the distribution of the independent sequence of  at an unknown point
at an unknown point  (change point) contained in the interval with length
(change point) contained in the interval with length  . Specifically, the change point model in the compound Poisson process can be formulated as
. Specifically, the change point model in the compound Poisson process can be formulated as

where  ,
,  and
and  are unknown means,
are unknown means,  is unknown variance of the normal distribution, and
is unknown variance of the normal distribution, and  ,
,  , and
, and  are unknown mean rates of biomarker occurrences in each subinterval. The goal of the study becomes to estimate the value of
are unknown mean rates of biomarker occurrences in each subinterval. The goal of the study becomes to estimate the value of  .
.
For illustration purpose, in the following Figure 1, we provide a scatter plot that represents a change in a sequence of data simulated from a compound Poisson process described above.

Simulated compound Poisson process data with one change: The upper panel is a plot of the simulated log ratio intensities (normally distributed) against the genomic positions, and the lower panel is a plot of the interval length against the corresponding genomic positions (distributed with Poisson).
2.2. A Bayesian Analysis for Locating the Change Point
The change-point model in the compound Poisson process described above can be viewed as a hypothesis testing problem. It tests the null hypothesis,  , of no change in the parameters of the sequence of random variables
, of no change in the parameters of the sequence of random variables  in subintervals with length
in subintervals with length 

versus the alternative hypothesis

The alternative hypothesis (7) above defines a change-point model. For this model, we propose a Bayesian approach for the estimate of  . Due to the requirement of occurrence in an interval, we only consider the search of the change when
. Due to the requirement of occurrence in an interval, we only consider the search of the change when  is between 2 and
is between 2 and  . We will obtain the posterior distribution of
. We will obtain the posterior distribution of  in the sequel. We first assume that the prior distribution of
in the sequel. We first assume that the prior distribution of  is taken as an noninformative prior
is taken as an noninformative prior

The following joint prior distribution is given for  ,
,  and
and 

and for the common variance  , the prior distribution is taken as
, the prior distribution is taken as

Under those assumptions, the likelihood function of  can be written as
can be written as

The joint posterior distribution of the parameters  ,
,  ,
,  ,
,  and
and  is then obtained as
is then obtained as

Integrating (12) above with respect to  ,
,  ,
,  and
and  , we found that the marginal posterior distribution of the interval
, we found that the marginal posterior distribution of the interval  that included the change point is proportional to
that included the change point is proportional to

for  , where the constants A, B, and C in (13) are obtained as
, where the constants A, B, and C in (13) are obtained as

The probability  in (13) is computed from the Poisson distribution with parameter
in (13) is computed from the Poisson distribution with parameter  for
for  according to the Poisson model under the alternative hypothesis
according to the Poisson model under the alternative hypothesis  (7), or namely
(7), or namely

In order to compute the probability given by (15), the occurrence rates  ,
,  , and
, and  can be estimated with the maximum likelihood estimator (MLE),
can be estimated with the maximum likelihood estimator (MLE),  ,
,  , and
, and  , in the subintervals of lengths
, in the subintervals of lengths  ,
,  , and
, and  , respectively. These MLEs are easily obtained as
, respectively. These MLEs are easily obtained as

With these MLEs, (15) becomes

Therefore, with the Poisson probabilities given by (17),  in (13) can be rewritten as
in (13) can be rewritten as

Finally, the marginal posterior distribution of the locus  is obtained as
is obtained as

where  is given in (18). The estimate of the change locus
is given in (18). The estimate of the change locus  is then given by
is then given by  such that the posterior distribution (19) attains its maximum at
such that the posterior distribution (19) attains its maximum at  , that is,
, that is,

Based on the above theoretical results, we provide the computational implementation of our approach in the next subsection.
2.3. Computational Implementation of the Bayesian Approach
To implement our above Bayesian approach to real data, it is necessary to define the number,  , of subintervals at first. Our numerical experiments show that the number,
, of subintervals at first. Our numerical experiments show that the number,  , of subintervals can be chosen such that each subinterval includes at least one observation (log ratio
, of subintervals can be chosen such that each subinterval includes at least one observation (log ratio  ) and at most 300 observations. The lengths,
) and at most 300 observations. The lengths,  ,
,  ,
, , and
, and  , of the subintervals can be chosen equally (in this case, the numbers of biomarkers contained in each subinterval are not equal). An easier option of choosing the length,
, of the subintervals can be chosen equally (in this case, the numbers of biomarkers contained in each subinterval are not equal). An easier option of choosing the length,  , for subinterval
, for subinterval  is to have each subinterval to contain the same number of observations. From a practical point of view, the number of subintervals,
is to have each subinterval to contain the same number of observations. From a practical point of view, the number of subintervals,  , and the size of each subinterval can also be defined by users according to their prior knowledge about their data.
, and the size of each subinterval can also be defined by users according to their prior knowledge about their data.
Although our approach was given for the single change-point model in compound Poisson process, it can be easily extended to the multiple change points (or aberrations) by using a sliding window approach [21, 22]. Sun et al. [21] have taken the sliding window sizes as 3 to 10 consecutive markers in their application. Our numerical experiments suggest that the sliding window of sizes ranging from 12 to 35 subintervals should be effective in searching for multiple changes in the aCGH data based on our proposed Bayesian approach. To avoid intermediate edge problems within each window, the two adjacent windows have to overlap. Many of such issues were also discussed in [22]. For the searching of multiple change points with the sliding window approach, a practical question is how to set the threshold value for the maximum posterior probabilities associated with all windows. In our application, we used the heuristic threshold of 0.5 (which is popular in probability sense) for the maximum posterior probabilities.
As a summary of our method, we give the following steps to implement our proposed Bayesian approach to the compound Poisson change-point model (Bayesian-CPCM).
-
(1)
If it is known that a chromosome has potentially one aberration region, calculate the posterior probability (19) and identify the locus
 according to (20).
according to (20). -
(2)
If there are multiple aberration regions on a chromosome or genome, choose a total of
 sliding windows with sizes ranging from 12 to 35 such that each window contains exactly one potential aberration. Denote these
sliding windows with sizes ranging from 12 to 35 such that each window contains exactly one potential aberration. Denote these  windows by
windows by  ,
, ,...,
,..., , where
, where 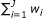 equals the total number of observations on the chromosome.
equals the total number of observations on the chromosome. -
(3)
For window
 , determine the number of subintervals
, determine the number of subintervals 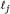 with lengths
with lengths  ,
, ,
,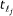 .
. -
(4)
Count the number of biomarkers,
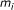 , in each subinterval with length
, in each subinterval with length 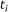 ,
,  .
. -
(5)
Compute the posterior probabilities for
 using (19), find the maximum of the posterior probability distribution. If the maximum posterior probability is larger than 0.5 (or larger than a selected threshold according to practice) at
using (19), find the maximum of the posterior probability distribution. If the maximum posterior probability is larger than 0.5 (or larger than a selected threshold according to practice) at  , then identify
, then identify  according to (20).
according to (20). -
(6)
Convert the identified change position
 into the actual biomarker position
into the actual biomarker position 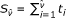 , and declare
, and declare 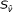 as the position on the chromosome at which the CNV has changed.
as the position on the chromosome at which the CNV has changed. -
(7)
Repeat steps 3–6 above for
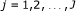 , where J is determined by the final window size and the final window size is determined at the value for which the posterior probabilities stabilize.
, where J is determined by the final window size and the final window size is determined at the value for which the posterior probabilities stabilize.
 according to (20).
according to (20). sliding windows with sizes ranging from 12 to 35 such that each window contains exactly one potential aberration. Denote these
sliding windows with sizes ranging from 12 to 35 such that each window contains exactly one potential aberration. Denote these  windows by
windows by  ,
, ,...,
,..., , where
, where 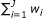 equals the total number of observations on the chromosome.
equals the total number of observations on the chromosome. , determine the number of subintervals
, determine the number of subintervals 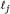 with lengths
with lengths  ,
, ,
,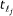 .
.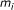 , in each subinterval with length
, in each subinterval with length 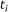 ,
,  .
. using (19), find the maximum of the posterior probability distribution. If the maximum posterior probability is larger than 0.5 (or larger than a selected threshold according to practice) at
using (19), find the maximum of the posterior probability distribution. If the maximum posterior probability is larger than 0.5 (or larger than a selected threshold according to practice) at  , then identify
, then identify  according to (20).
according to (20). into the actual biomarker position
into the actual biomarker position 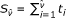 , and declare
, and declare 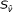 as the position on the chromosome at which the CNV has changed.
as the position on the chromosome at which the CNV has changed.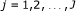 , where J is determined by the final window size and the final window size is determined at the value for which the posterior probabilities stabilize.
, where J is determined by the final window size and the final window size is determined at the value for which the posterior probabilities stabilize.The Matlab code of the Bayesian-CPCM approach has been written and is available upon readers' request.
3. Results
3.1. Simulation Results
The proposed method provides a theoretic framework of detecting CNVs using both biomarker positions and log-intensity ratios. Since there is no suitable metric that can be used to compare the proposed approach with all existing algorithms, we carried simulation studies based on a commonly used approach for evaluating the estimation of a change point. We simulated sequences as independent normal distributions with moderate sample size  (the sequence size) of 12, 20, 32, 40, 80, and 120 for the scenarios of the changes being located at the front (the
(the sequence size) of 12, 20, 32, 40, 80, and 120 for the scenarios of the changes being located at the front (the  th observation), at the center (the
th observation), at the center (the  th observation), and at the end (the
th observation), and at the end (the  th observation) of the respective sequence. For the choices of the mean and variance parameters before and after the change location, we consider the specific features of the real aCGH data. Using data from the fibroblast cell lines as benchmarks, we observed that the segments before and after a detected change point mostly have mean difference ranging from .36 to .7 (or larger), and a standard deviation difference ranging mostly from .05 to .2. We, therefore, investigated the cases when the mean and the standard deviation are within the above-mentioned ranges. Due to the page limit of the paper, we only report part of the simulation results in Table 1. In Table 1,
th observation) of the respective sequence. For the choices of the mean and variance parameters before and after the change location, we consider the specific features of the real aCGH data. Using data from the fibroblast cell lines as benchmarks, we observed that the segments before and after a detected change point mostly have mean difference ranging from .36 to .7 (or larger), and a standard deviation difference ranging mostly from .05 to .2. We, therefore, investigated the cases when the mean and the standard deviation are within the above-mentioned ranges. Due to the page limit of the paper, we only report part of the simulation results in Table 1. In Table 1,  denotes the true change location;
denotes the true change location;  is the estimated change location according to (20);
is the estimated change location according to (20);  represents the relative frequency that the estimated location
represents the relative frequency that the estimated location  equals to the true location
equals to the true location  ; and
; and  is the mean squared error of the location estimator. Each simulation is carried out 1,000 times.
is the mean squared error of the location estimator. Each simulation is carried out 1,000 times.
The simulation results given in Table 1 indicate that the derived posterior probability (19) can identify changes in the front, the center and the end of the sequence, respectively, with very high certainty—at least 97% for sample sizes of 20 or larger. The average of the estimated locations is remarkably close to the true change locus with very small MSE. The proposed method can be confidently applied to the identification of DNA copy number changes.
3.2. Applications to aCGH Datasets on 9 Fibroblast Cell lines
Several aCGH experiments were performed on 15 fibroblast cell lines and the normalized averages of the  (based on triplicate) along positions on each chromosome were available at the following website [23]: http://www.nature.com/ng/journal/v29/n3/full/ng754.html. For the missing values in the log ratio values, we imputed 0 into the original data. The DNA copy number alterations in each of the 15 fibroblast cell lines were verified by karyotyping [23]. Therefore, these 15 fibroblast cell lines aCGH datasets can be used as benchmark datasets to test our methods.
(based on triplicate) along positions on each chromosome were available at the following website [23]: http://www.nature.com/ng/journal/v29/n3/full/ng754.html. For the missing values in the log ratio values, we imputed 0 into the original data. The DNA copy number alterations in each of the 15 fibroblast cell lines were verified by karyotyping [23]. Therefore, these 15 fibroblast cell lines aCGH datasets can be used as benchmark datasets to test our methods.
For the 9 fibroblast cell lines analyzed in many followup papers of [23], we also used our posterior probabilities (19) to locate the locus (or loci) on those chromosomes where the alterations had been identified. It turned out that our method can identify the locus (or loci) of the DNA copy number alterations that are exactly corresponding to the karyotyping results [23]. The CNVs found by our proposed Bayesian approach (with sliding windows when appropriate) are summarized in the following Tables 2 and 3.
According to the posterior probability (19), we found that there was one copy number change on chromosome 5 of the cell line GM01535, chromosomes 9 and 14 of the cell line GM01750, chromosomes 3 and 9 of the cell line GM03563, chromosome 7 of the cell line GM07081, and chromosomes 1 and 4 of the cell line GM13330. No false positives were found on these chromosomes with the threshold of 0.5 for the maximum posterior probability (20). These findings are consistent with the karyotyping result of Snijders et al. [23]. In Figures 2 and 3, we give the scatter plots of the aCGH data of Chromosome 3 of GM03563, and of Chromosome 7 of GM07081, along with their respective posterior probability distributions. The peak posterior indicated a change at that genomic locus. The beginning point after which the corresponding log ratio values are increased is circled as red.

Chromosome 3 of GM03563 [23] with identified change locus and the posterior probability distribution: A red circle indicates a significant DNA copy number change point such that the segment before this red circle (inclusive of the red circle) is different from the successor segment after the red circle (exclusive of the red circle)

Chromosome 7 of GM07081 [23] with identified change locus and the posterior probability distribution: A red circle indicates a significant DNA copy number change point such that the segment before this red circle (inclusive of the red circle) is different from the successor segment after the red circle (exclusive of the red circle).
Our posterior probability function of (20) combined with the sliding window approach signals two or more possible copy number changes on chromosome 6 of GM01524, chromosome 8 of GM03134, chromosomes 10 and 11 of GM05296, and chromosome 17 of GM13031. These results were given in Table 2. Figures 4 and 5 give the findings on Chromosome 6 of GM01524 and Chromosome 17 of GM13031, respectively, with a sliding window approach used. These findings are again consistent with the karyotyping result of [23].

Chromosome 6 of GM01524 [23] with identified change loci (indicated by red arrows) and the posterior probability distributions with a window size of 20

Chromosome 17 of GM13031 [23] with identified change loci (indicated by red arrows, while the green arrow indicates a false positive) and the posterior probability distributions with a window size of 20.
3.3. Comparison of the Performances of the Proposed Bayesian-CPCM with CBS on the Fibroblast Cell-Lines Datasets
There are many approaches (computational or statistical) now available for analyzing aCGH data in the relative literature. But many of those approaches, especially CBS [4], have targeted on modeling the log ratio intensity in aCGH data. Now, in this paper, we have used a new concept to model both the gene position and the log ratio intensity in aCGH data. That is, the most distinct feature of the proposed Bayesian-CPCM approach, among other existing methods in the literature, is its usage of the information of the gene positions (hence gene distances) and the log ratio intensities in the model.
Although there is no suitable metric that can be used to compare all the existing methods for CNV data analysis, we used the specificity and sensitivity as comparison metric to evaluate the performance of our proposed method with one of the most popularly used CBS method. The comparison results are given in the following Table 4. In Table 4, "Yes" means the change was found by the specific method (CBS or Bayesian-CPCM) for the known alteration verified by spectral karyotyping in Snijders et al. [23] on the specific chromosome in the cell line at the given  level (for the case of using CBS or MVCM) or with maximum posterior probability larger than 0.5 (for the case of using Bayesian-CPCM), "No" means the change was not found by a specific method, but was identified by spectral karyotyping; and "Number of false positives" gives the number of changes found by the specific method for a cell line while there were no known alterations actually found by spectral karyotyping [4, 23].
level (for the case of using CBS or MVCM) or with maximum posterior probability larger than 0.5 (for the case of using Bayesian-CPCM), "No" means the change was not found by a specific method, but was identified by spectral karyotyping; and "Number of false positives" gives the number of changes found by the specific method for a cell line while there were no known alterations actually found by spectral karyotyping [4, 23].
From Table 4, it is evident that the new Bayesian-CPCM approach can detect the CNV regions with highest specificities and sensitivities. The false positives of the Bayesian-CPCM on two of the chromosomes are due to outliers and noise in the original data.
It is worth noting that the CNV or aberration regions in these 9 fibroblast cell lines that were found using our proposed Bayesian-CPCM approach are also consistent with those identified in Olshen et al. [4], Chen and Wang [19], Venkatraman and Olshen [24]. However, our new approach, Bayesian-CPCM, neither involve heavy computations as that of CBS algorithm in Olshen et al. [4], nor any asymptotic distribution as required in our earlier work [19].
4. Conclusion
A Bayesian approach for identifying CNVs in aCGH profile modeled by a compound Poisson process is proposed in this paper. Theoretical results of the Bayesian analysis are obtained and the algorithm has been implemented with Matlab. Applications of the proposed method to several aCGH data sets have demonstrated its effectiveness. Extensive simulation results indicate that the proposed method can work effectively for various cases. The most distinct feature of the proposed Bayesian-CPCM approach, when compared with existing methods in the literature, is its use of both biomarker positions (hence distances) and the log-intensity ratio information in the model. Another important aspect of the proposed approach is that it characterizes the posterior probability of the loci being a CNV. With the common knowledge of probability, the users can easily judge if there is a CNV at a locus by using the posterior probability together with their biological knowledge.
There are many computational and statistical approaches now available for analyzing aCGH data in the literature. But those approaches, especially the CBS of Olshen et al. [4] and MVCM of Chen and Wang [19], are all targeted on modeling the log ratio in aCGH data. In this paper, we have used a new approach to model both the biomarker position and the log ratio intensity in aCGH data. In other words, the most distinct feature of the proposed Bayesian-CPCM approach, among other existing methods, is the use of both biomarker position information (hence distances) and the log-intensity ratios in the model. The size of the sliding window is very important in search multiple change points in a whole sequence. The criterion of choosing the optimal window size remains to be done in the future.
References
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, González JR, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME: Global variation in copy number in the human genome. Nature 2006, 444(7118):444-454. 10.1038/nature05329
Pinkel D, Seagraves R, Sudar D, et al.: High resolution analysis of DNA copy number variation usingcomparative genomic hybridization to microarrays. Nature Genetics 1998, 20: 207-211. 10.1038/2524
Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO: Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nature Genetics 1999, 23(1):41-46. 10.1038/12640
Olshen AB, Venkatraman ES, Lucito R, Wigler M: Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5(4):557-572. 10.1093/biostatistics/kxh008
Levin AM, Ghosh D, Cho KR, Kardia SLR: A model-based scan statistic for identifying extreme chromosomal regions of gene expression in human tumors. Bioinformatics 2005, 21(12):2867-2874. 10.1093/bioinformatics/bti417
Hodgson G, Hager JH, Volik S, Hariono S, Wernick M, Moore D, Nowak N, Albertson DG, Pinkel D, Collins C, Hanahan D, Gray JW: Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas. Nature Genetics 2001, 29: 459-464. 10.1038/ng771
Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale A-L, Brown PO: Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proceedings of the National Academy of Sciences of the United States of America 2002, 99(20):12963-12968. 10.1073/pnas.162471999
Hupé P, Stransky N, Thiery J-P, Radvanyi F, Barillot E: Analysis of array CGH data: from signal ratio to gain and loss of DNA regions. Bioinformatics 2004, 20(18):3413-3422. 10.1093/bioinformatics/bth418
Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, Jänne PA, Girard L, Minna J, Christiani D, Li C, Sellers WR, Meyerson M: Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Research 2005, 65(13):5561-5570. 10.1158/0008-5472.CAN-04-4603
Picard F, Robin S, Lavielle M, Vaisse C, Daudin J-J: A statistical approach for array CGH data analysis. BMC Bioinformatics 2005., 6, article 27:
Lai WR, Johnson MD, Kucherlapati R, Park PJ: Comparative analysis of algorithms for identifying amplifications and deletions in array CGH data. Bioinformatics 2005, 21(19):3763-3770. 10.1093/bioinformatics/bti611
Nannya Y, Sanada M, Nakazaki K, et al.: A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Research 2005, 65: 6071-6079. 10.1158/0008-5472.CAN-05-0465
Price TS, Regan R, Mott R, Hedman Å, Honey B, Daniels RJ, Smith L, Greenfield A, Tiganescu A, Buckle V, Ventress N, Ayyub H, Salhan A, Pedraza-Diaz S, Broxholme J, Ragoussis J, Higgs DR, Flint J, Knight SJL: SW-ARRAY: a dynamic programming solution for the identification of copy-number changes in genomic DNA using array comparative genome hybridization data. Nucleic Acids Research 2005, 33(11):3455-3464. 10.1093/nar/gki643
Shah SP, Xuan X, DeLeeuw RJ, Khojasteh M, Lam WL, Ng R, Murphy KP: Integrating copy number polymorphisms into array CGH analysis using a robust HMM. Bioinformatics 2006, 22(14):e431-e439. 10.1093/bioinformatics/btl238
Yu T, Ye H, Sun W, Li K-C, Chen Z, Jacobs S, Bailey DK, Wong DT, Zhou X: A forward-backward fragment assembling algorithm for the identification of genomic amplification and deletion breakpoints using high-density single nucleotide polymorphism (SNP) array. BMC Bioinformatics 2007., 8, article 145:
Rueda OM, Díaz-Uriarte R: Flexible and accurate detection of genomic copy-number changes from aCGH. PLoS Computational Biology 2007, 3(6):1115-1122.
Pique-Regi R, Monso-Varona J, Ortega A, Seeger RC, Triche TJ, Asgharzadeh S: Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics 2008, 24(3):309-318. 10.1093/bioinformatics/btm601
Rancoita PMV, Hutter M, Bertoni F, Kwee I: Bayesian DNA copy number analysis. BMC Bioinformatics 2009., 10, article 10:
Chen J, Wang Y-P: A statistical change point model approach for the detection of DNA copy number variations in array CGH data. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2009, 6: 529-541.
Daruwala R-S, Rudra A, Ostrer H, Lucito R, Wigler M, Mishra B: A versatile statistical analysis algorithm to detect genome copy number variation. Proceedings of the National Academy of Sciences of the United States of America 2004, 101(46):16292-16297. 10.1073/pnas.0407247101
Sun YV, Levin AM, Boerwinkle E, Robertson H, Kardia SLR: A scan statistic for identifying chromosomal patterns of SNP association. Genetic Epidemiology 2006, 30(7):627-635. 10.1002/gepi.20173
Ramensky VE, Makeev VJu, Roytberg MA, Tumanyan VG: DNA segmentation through the Bayesian approach. Journal of Computational Biology 2000, 7(1-2):215-231. 10.1089/10665270050081487
Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, Law S, Myambo K, Palmer J, Ylstra B, Yue JP, Gray JW, Jain AN, Pinkel D, Albertson DG: Assembly of microarrays for genome-wide measurement of DNA copy number. Nature Genetics 2001, 29(3):263-264. 10.1038/ng754
Venkatraman ES, Olshen AB: A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23(6):657-663. 10.1093/bioinformatics/btl646
Acknowledgments
Part of the paper was done while A. Yi iter was on leave from Hacettepe University and was a visiting scholar at the University of Missouri-Kansas City with financial support provided by the Scientific and Technological Research Council of Turkey (TUBITAK). J. Chen was supported in part by a 2009 University of Missouri Research Board (UMRB) research Grant. H.-W. Deng was partially supported by grants from NIH (nos. P50 AR055081, R01AR050496, R01AR45349, and R01AG026564) and by Dickson/Missouri endowment.
iter was on leave from Hacettepe University and was a visiting scholar at the University of Missouri-Kansas City with financial support provided by the Scientific and Technological Research Council of Turkey (TUBITAK). J. Chen was supported in part by a 2009 University of Missouri Research Board (UMRB) research Grant. H.-W. Deng was partially supported by grants from NIH (nos. P50 AR055081, R01AR050496, R01AR45349, and R01AG026564) and by Dickson/Missouri endowment.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, J., Yiğiter, A., Wang, YP. et al. A Bayesian Analysis for Identifying DNA Copy Number Variations Using a Compound Poisson Process. J Bioinform Sys Biology 2010, 268513 (2010). https://doi.org/10.1155/2010/268513
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1155/2010/268513